INTRODUCTION
Campomelic dysplasia (CD) is a rare genetic disease that belongs to a group of lethal skeletal dysplasias. It is inherited as an autosomal dominant disorder. The term campomelia is derived from the Greek word campo (or campto), meaning bent limb [1] and is characterized by bowing of the long bones. Campomelia also affects the development of the skeleton, reproductive system, and other organ systems. This disorder is often life-threatening in newborns because it causes respiratory distress. Another characteristic of this disorder is that most patients manifest male-to-female sex reversal. Further, some patients with the male karyotype (XY) have female or ambiguous genitalia [2] caused by a mutation in SOX9, a member of the SOX (SRY-related HMG box) gene family. We here describe such a case involving a frameshift mutation in SOX9 that has not previously been reported.
CASE REPORT
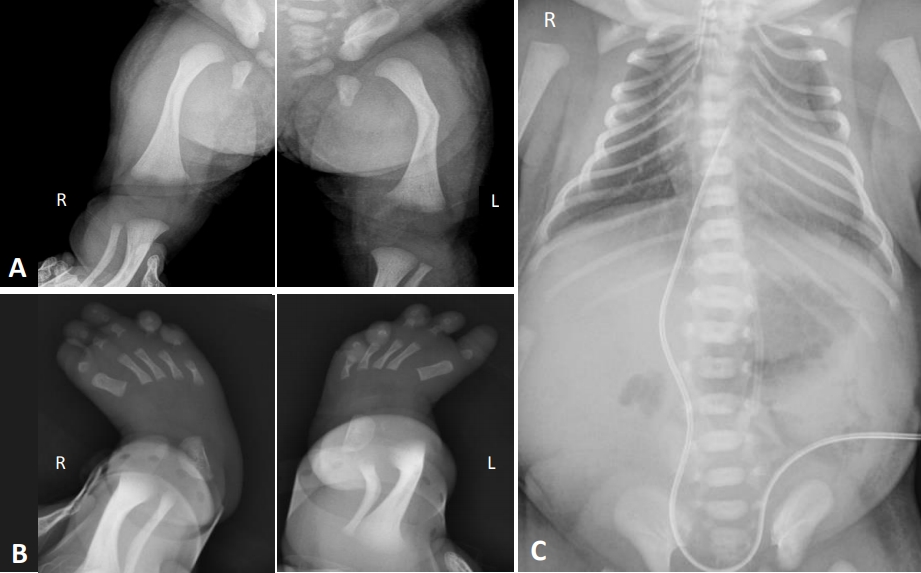
Our CD patient was a female born in 2017 by cesarean section after 37 weeks of gestation to a 29-year-old mother. She was the first child of a healthy, non-consanguineous couple. Her mother was gravida 0, para 0. There was no history of skeletal abnormalities on both sides of the family. A prenatal ultrasound demonstrated angulation and bowing of both femurs, contracture of her left knee joint, malalignment of both toes, micrognathia, and female external genitalia. A 46, XY karyotype was confirmed by amniocentesis. At birth, her Apgar scores were 2 and 5, respectively, and endotracheal intubation was performed due to respiratory distress in the delivery room. Her birth measurements were as follows: weight, 3.1 kg (50th to 90th percentile); length, 45 cm (10th percentile); and head circumference, 38 cm (>97th percentile). Her physical features included micrognathia, macroglossia, a cleft palate, a flattened nasal bridge, low set ears, a small thoracic cage, short limbs, anterolateral femoral bowing, clubbed feet, and normal female external genitalia. Laboratory blood tests were normal. A postnatal skeletal radiograph confirmed the bowing of both femurs (Figure 1A) and clubbed feet (Figure 1B), and also revealed a hypoplastic scapula, 11 pairs of ribs, a bell-shaped narrow thorax, and scoliosis (Figure 1C). Echocardiography further revealed a patent foramen ovale without anomalies. Cerebral ultrasonography on the 2nd day post-birth revealed mildly prominent lateral ventricles. Abdominal and genital ultrasonography revealed a vagina and uterus but no ovaries or testicles. The result of brainstem auditory evoked potential was both referred.
We obtained informed consent from the parents to screen for genetic mutations and we isolated peripheral blood genomic DNA for this purpose. Nucleotide sequence analysis revealed a heterozygous c.235delC deletion mutation in exon 1 of the SOX9 gene, which we identified as a previously unreported p.Gln79Argfs*31 frameshift mutation (Figure 2).
Ventilator weaning in this patient failed due to respiratory distress. A tracheal ring and mild tracheomalacia were detected by bronchoscopy. A tracheostomy was performed at 24 days of age. A subsequent video fluoroscopic swallowing study indicated severe dysphagia, which resulted in nasogastric tube feeding. There was one seizure event during the hospitalization period. There were no specific findings from brain magnetic resonance imaging or electroencephalography. Anticonvulsant medications were eventually discontinued since the symptoms had abated. She was discharged 83 days after birth with a home ventilator.
The patient was again hospitalized due to pneumonia at 10 months of age. Palatoplasty and pressure equalization tube insertion were performed at 12 months of age. A Bayley Scales of Infant and Toddler Development-II evaluation was performed when she was 14 months old. The mental development and psychomotor development indices were both less than 50. The mental and motor developmental ages according to the raw scores were 3 and 4 months, respectively, indicating overall developmental delay. The number of months according to the scales revealed that she had the cognitive ability of a 3-month-old, the language understanding and expression of an infant less than 1-month-old, the social development of a 2 to 3-month-old, the gross motor skills of an infant less than 1-month-old, and the fine motor skills of a baby of 3 to 4 months of age. At 2 years, her weight, height, and head circumference were 9.4 kg (<3rd percentile), 72.2 cm (<3rd percentile), and 47.5 cm (50th percentile), respectively. She remained completely dependent on others due to her developmental delays. She received a left cochlear implant when she was 2 years old. Although our patient was in a critical condition as a newborn due to respiratory distress, she was eventually maintained in a stable condition through the use of a home ventilator. She is 3 years of age at the time of writing and is undergoing rehabilitation treatment for her developmental delays and we conduct regular follow-ups in an outpatient setting.
DISCUSSION
CD is a rare genetic disease but should be considered if there are certain characteristic manifestations in neonates such as macrocephaly with a flattened face and nasal bridge, low-set ears (often with associated deafness), hypertelorism, a long philtrum, micrognathia, and skeletal dysplasia. The skeletal features of CD include bowing of both tibia, angulated femurs, hypoplastic scapula, 11 pairs of ribs, a bell-shaped narrow thorax, and characteristic pretibial skin dimples [2]. This disorder can often be life-threatening due to the onset of respiratory distress during the neonatal period. If CD is clinically suspected, the SOX9 gene should be sequenced to identify possible mutations.
SOX9 is a member of the family of SRY-related high-mobilitygroup box (SOX) transcription factors [3]. The SOX9 gene is located on the long arm of human chromosome 17. The SOX9 protein HMG box domain shares 71% similarity with the SRY HMG box and has a proline and glutamine rich region in its activation domain [4]. SOX9 mutations are associated with skeletal dysplasia and sex-reversal phenotypes. However, their precise mechanistic relationship with sex-reversal phenotypes has not been established [5]. SOX9 regulates chondrogenesis and the expression of cartilage matrix genes in the early stages of development [6]. It binds to the chondrocyte-specific enhancer of the COL2A1 gene, which encodes type II collagen, the main cartilage matrix protein, and thereby activates it in these cells. SOX9 is also a transcription factor that regulates the differentiation of cartilage cells. Low levels of SOX9 lead to a decrease in COL2A1 expression and thereby cause skeletal abnormalities [7,8].
In addition to chondrogenesis, the SOX9 gene plays a role in sex determination and gonadogenesis in the early stages of development. SRY may directly or indirectly control the timing and cell-type specificity of SOX9 expression [9]. X9 regulates the differentiation of Sertoli cells and some SOX9 mutations may cause male-to-female sex reversal [2]. Chondrogenesis is possible only when SOX9 binds cooperatively as a dimer. SOX9 mutations do not always cause sex reversal because the mutant proteins can operate as monomers and bind to the regulatory regions of the sex-determining gene SF1.
Four classes of heterozygous SOX9 mutations were identified Crompreviously by Bernard et al. [10]: (1) amino acid substitutions in the HMG domain; (2) truncations or frameshifts that alter the C terminus; (3) mutations at splice junctions; and (4) chromosomal translocations. In our current case, the frameshift we identified caused a deletion of codon 235 in exon 1 of SOX9 as a heterozygous, c.235delC, variation.
To date, two cases of CD have been genetically confirmed in Korea [11,12]; however, the p.Gln79Argfs*31 frameshift deletion mutation in our current study has never been reported. This premature termination of the SOX9 protein results in a loss of function, leading to skeletal dysplasia and sexual reversal. As in this case, two previous studies from Korea have reported severe clinical courses: in one case, the patient died of respiratory failure after 4 months, and, at the time of writing, the other patient was 40 months old [11,12].
Unlike in Korea, foreign cases are often found who have survived for a long time or have been diagnosed with old age. According to Meyer et al. [13], there is no correlation between the type and position of mutations within the SOX9 gene among phenotypes. However, in other cases where the CD patients survived, most of the patients exhibited milder symptoms, and it should be noted that the difference determines the severity of symptoms. However, correlations of some degree are observed in those with: (1) chromosomal rearrangement and (2) mosaicism of the SOX9 mutation [14,15]. De novo translocations or inversions with breakpoints upstream of SOX9 are more likely to lead to the removal of one or more cis-regulatory elements [15]. According to Mansour et al. [14], the father of the patient also has the same SOX9 mutation, but his mutation was clinically silent. These findings were associated with better survival or diagnosis at an older age.
In conclusion, we here report a novel frameshift mutation in the SOX9 gene of a female with a 46, XY karyotype who was born with CD and male-to-female sex reversal. CD is a very rare and often fatal genetic disorder; however, the early diagnosis and management of related complications in affected children can prevent the deterioration of their clinical condition and improve their quality of life.