INTRODUCTION
14q12q13 Microdeletion syndrome is a major chromosome anomaly associated with neurodevelopmental delay and learning disability, as well as microcephaly, failure to thrive, seizure, and abnormal development of part of the brain [1]. The symptoms may vary depending on the gene deleted. Deletion of the Forkhead box protein G1 (FOXG1) gene manifests as a neurodevelopmental disability or severe developmental delay, while a deletion of NK2 homeobox 1 (NKX2-1) has been associated with the brain-lung-thyroid syndrome and has thus been reported to result in severe pulmonary insufficiency and hypothyroidism [2].
While this microdeletion syndrome is associated with severe clinical manifestations, it cannot be detected with routine chromosome analysis and is thus mistaken for another disease or has a delayed diagnosed. For this reason, various tests are continually performed to determine a specific diagnosis, and deciding upon the direction of treatment may be challenging. Hence, the United States implemented chromosomal microarray analysis as the primary diagnostic test for patients with various congenital anomalies or developmental delay 10 years ago, and such a trend has begun to emerge in Korea in recent years [3,4].
The present case is of an infant who underwent various tests including routine karyotyping for hypoplasia of the corpus callosum, reduced muscle tension, seizure, and hypothyroidism but could not be diagnosed at the neonatal intensive care unit. However, while undergoing treatment for seizure and transient hypothyroidism, the patient showed clinical worsening of neurodevelopment. The patient was then evaluated for a possible genetic syndrome and subsequently diagnosed with 14q12q13.3 microdeletion syndrome by chromosomal microarray analysis. The infant had a large deletion spanning 11.5 Mb and is the first case of 14q12q13.3 microdeletion with diverse and severe symptoms to be reported in Korea.
CASE REPORT
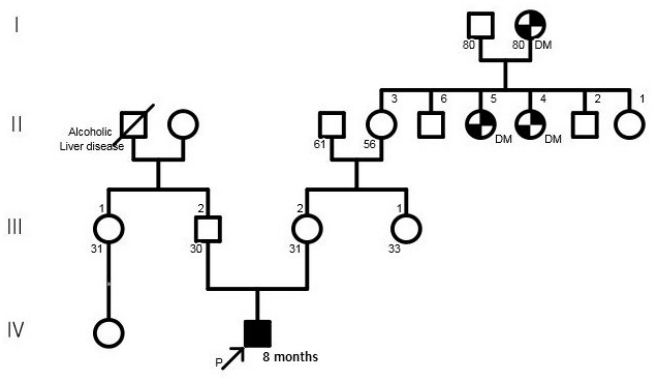
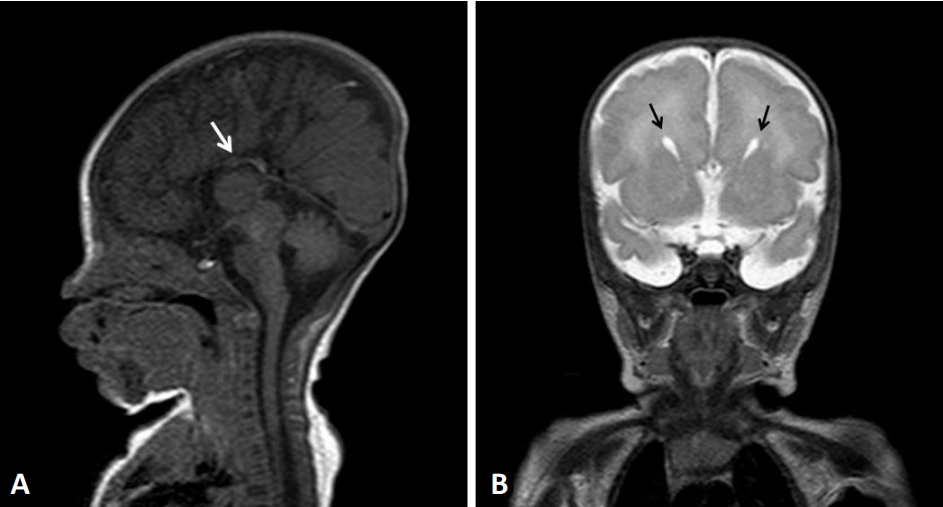
A 30-year-old pregnant woman with obstetrical history of gravida 1, parity 0 delivered a 1.93 kg boy at 36 weeks and 4 days of gestation due to fetal distress through vaginal delivery. She had been treated for premature uterine contractions from 25 weeks of gestation. Although no abnormalities were found during prenatal care, the amniotic fluid index checked on 35 weeks of gestation was 5.1 cm, based on which oligohydramnios was suspected, and the fetus showed intrauterine growth delay. The results of placental biopsy conducted postpartum were normal. The father of the baby was 29 years of age, without any notable family or medical history (Figure 1). The baby’s Apgar score at birth was 7 at 1 minute and 9 at 5 minutes. The baby showed symmetrical intrauterine growth retardation, with a birth weight below the 3rd percentile and a head circumference (31 cm) and height (44 cm) below the 10th percentile. The baby had small capillary hemangiomas at the center of the forehead and posterior scalp, a simian line on the right hand, and slightly diminished muscle tone. He had stridor and tachypnea requiring oxygen via a nasal cannula, and nasogastric tube feeding was initiated due to tachypnea. The first blood test taken on the day of the birth showed a white blood cell count of 7,600/mm3, a platelet count of 116,000/mm3, and a C-reactive protein (CRP) level of 3.5 mg/dL, raising suspicion for early sepsis; thus, antibiotic therapy was initiated. On hospital day (HD) 6, the platelet count dropped to 35,000/mm3. The prothrombin time (PT), activated partial thromboplastin time (aPTT), and international normalized ratio (INR) were 16.3 seconds, 46.9 seconds, and 1.30, respectively, indicating slightly increased PT with aPTT and INR within the normal ranges. Antithrombin III was 46%, D-dimer/fibrin degradation product tests were positive, and systemic bruising was noted. Therefore, platelet concentrate and fresh frozen plasma were transfused, and antithrombin was administered. On HD 12, no bacteria were identified in the blood culture, the platelet count increased to 104,000/mm3, and the CRP level improved, leading to the discontinuation of antibiotic therapy. Tests for congenital infection, such as cytomegalovirus urine polymerase chain reaction and TORCH immunoglobulin M level, were normal. Oral feeding was attempted on HD 6; however, the baby was not feeding well, because he was not strong enough to suck the bottle. Therefore, central venous parenteral nutrition was started. On HD 15, the baby had a seizure, with stiffening and shaking of the extremities and was placed on a ventilator due to repeated episodes of apnea and reduced oxygen saturation. An electroencephalogram (EEG) showed moderate encephalopathy with frequent negative sharp waves on both central regions. Seizures were poorly controlled with phenobarbital; therefore, topiramate was administered. Brain computed tomography performed on HD 18 revealed suspected hypoplasia of the corpus callosum. Ventilator use was discontinued on HD 22 after the baby’s seizures ceased and his breathing stabilized. Hypoplasia of the corpus callosum was confirmed on brain magnetic resonance imaging (MRI), with no additional anomalies discovered (Figure 2). As the baby had symmetrical intrauterine growth retardation, simian line, hypoplasia of the corpus callosum, and demonstrated muscle weakness and seizure, routine chromosomal karyotyping was performed using peripheral blood. This confirmed the baby to be a male with normal 46 XY chromosomes. Echocardiography, renal ultrasonography, and eye examination performed to reveal any other anomalies showed normal findings, and diagnostic auditory brainstem response confirmed normal hearing in the left ear but mild hearing loss with a hearing threshold of 40 dB in the right ear.
While the initial thyroid stimulating hormone (TSH) level on the neonatal metabolic screening test (NST) was normal, a second NST taken after the seizure showed an elevated TSH level of 17.1 μIU/mL. On HD 20, hypothyroidism was diagnosed with a TSH level of 37.80 μIU/mL, free thyroxine level of 0.92 ng/dL, and triiodothyronine level of 1.06 ng/dL. The thyroglobulin level was elevated to 586.59 ng/mL, and anti-thyroglobulin antibody (Ab) and anti-thyroid peroxidase (anti-TPO) Ab were negative. Thyroid ultrasonographic findings were unremarkable. Under the diagnosis of hypothyroidism, the baby was administered 10 μg/kg of levothyroxine, after which the thyroid function test results were normal.
The baby gained weight to 2.92 kg with increased oral feeding, and the seizures were controlled. The baby was diagnosed with seizure and transient hypothyroidism and was discharged on HD 54 with continued phenobarbital, topiramate, and levothyroxine therapy. At this time, the baby continued to show muscle weakness equivalent to a level 4 grading using the Manual Muscle Testing method and was scheduled for rehabilitation and followup in the outpatient clinic.
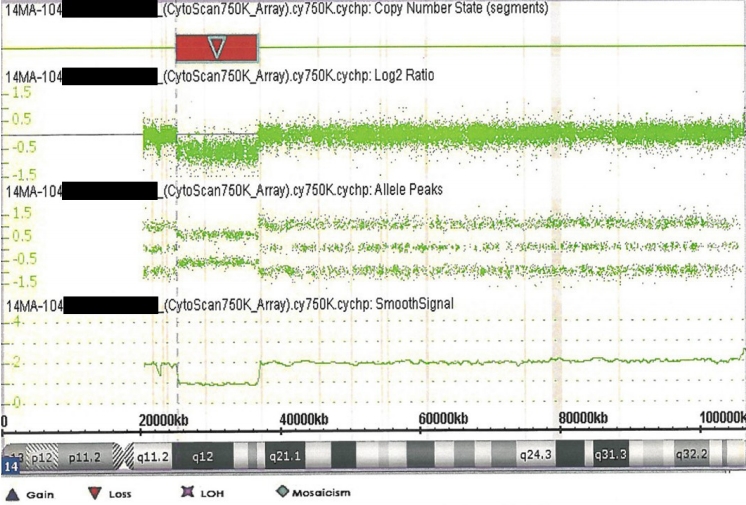
Two days after discharge, the baby was brought to the emergency department for poor feeding and lethargy. He was administered intravenous fluid therapy and discharged. Subsequently, he was brought to the hospital several times due to irritability and repeated hypertonia until 4 months of age, and each time, conservative treatment was conducted. He had no seizures and a normal EEG at 4 months of age; therefore, anticonvulsants were ceased. At 5 months of age, the baby was diagnosed with failure to thrive with a weight of 4.6 kg (<1st percentile), height of 50 cm (<1st percentile), head circumference of 39 cm (<3rd percentile), hypertonia, sleep disturbance with irritability, and constipation. The baby developed some neck strength but could not make eye contact. On the Bayley Scales of infant and toddler development, 3rd edition, at 6 months of age his cognition composite score was 55 (0.1st percentile), language composite score was 59 (0.3st percentile), and motor composite score was 46 (<0.1st percentile), which revealed severe developmental delay equivalent to the development of a 1-month-old. At 8 months of age, the baby was referred to a Genetic Counseling Clinic and underwent genetic evaluation including pedigree analysis, which was essentially negative except diabetic mellitus in maternal great aunts, and chromosomal microarray testing. The test detected an in11.5 Mb deletion in chromosome 14q [14q12q13.3 (25,363,718-36,872,996)×1] (Figure 3). At 19 months of age, he again began to have seizures with the arms and legs flexed. Valproate therapy was initiated followed by topiramate, when valproate alone could not control the seizures. It was difficult to increase the amount of feeding, as he was able to feed using a bottle but had difficulty swallowing. Further, he was frequently given antibiotic therapy due to repeated respiratory infections. The Denver developmental screening test conducted at 25 months of age revealed that the child was at 3 months for large motor development, 2 months for personal-social, and 3 months for language development. Thyroid function tests revealed normal findings until 36 months of age, and levothyroxine was stopped. However, after 2 weeks, his TSH level rose again to 18.34 μIU/mL; therefore, levothyroxine therapy was resumed, resulting in a level within the normal range. At 41 months of age, the baby’s weight was 7.7 kg (<1st percentile), height was 72 cm (<1st percentile), and head circumference was 44 cm (<1st percentile). The child was still only able to feed from a bottle, with weight gain stalled, so enteric nutrients were added for calorie supplementation. Pharmacological therapy for seizures and hypothyroidism was continued. His large motor and language development were equivalent to those of a 3-monthold, where he developed neck strength, was able to lift his head 45 degrees when placed on his stomach, and was able to produce only meaningless sounds.
DISCUSSION
Approximately 2% of neonates born worldwide have a major anomaly [5], 10% of which are known to result from a chromosome abnormality [6]. The incidence is gradually on the rise due to the increasing age of mothers at childbirth, and the increase in in vitro fertilization [7]. The banding karyotyping technique is useful for confirming abnormalities resulting from chromosome translocations, inversions, ring chromosomes, marker chromosomes, and deletions [8,9]. However, more than half of neonatal chromosomal abnormalities cannot be detected by physical examination or banding karyotyping [8,10,11], and these are cases in which a sex chromosomal abnormality has not yet manifested or cases of microdeletion syndrome that are discovered via chromosomal microarray analysis. Microdeletion syndrome refers to cases in which the chromosomal defect is smaller than 5 Mb, which is difficult to detect even with a high-resolution banding technique. Chromosomal microarray analysis has a higher sensitivity to microdeletions, subtelomeres, and pericentric regions than routine karyotyping and is thus known to be conducive to the diagnosis of microdeletion or microduplication syndrome [12,13].
The present case is of an infant with intrauterine growth retardation, microcephaly, muscle weakness, seizures, hypothyroidism, and hypoplasia of the corpus callosum who showed severe growth and neurodevelopmental abnormalities during follow-up and was diagnosed with 14q12q13.3 deletion via chromosomal microarray analysis.
The patient in this case had 14q12q13.3 deletion, which includes the FOXG1, COCHAP4S1, CFL2, PSMA6, NFKBIA, and NKX2-1 genes (Figure 4). Accordingly, he showed microcephaly, structural anomaly of the corpus callosum, seizures, and severe developmental delays, which are observed with FOXG1 gene anomalies [14,15], along with neonatal hypotonicity that progresses to hypertonia, developmental delay, and seizures, which are observed with AP4S1 mutation [16].
Furthermore, while the baby had normal findings on the initial NST after birth, a thyroid function test performed after the onset of seizures confirmed hypothyroidism. The NKX2-1 gene is located on 14q13.3 and is involved in the formation of homeobox protein Nkx-2.1; therefore, this gene affects thyroid hormone production. In the present case, we speculate that the patient’s hypothyroidism is caused by the abnormality of this gene [17].
The NFKBIA gene is associated with ectodermal dysplasia; affects hair, teeth, and sweat gland development; and results in T cell immunodeficiency [18]. The child in the present case had no dental development and experienced repeated respiratory infections. COCH, CFL2, and PSMA6 are associated with hearing abnormality, nemaline myopathy, and ophthalmologic diseases, respectively, and the patient in the present case did not show these symptoms.
Existing descriptions of 14q12q13.3 microdeletion are those reported by Fonseca et al. [19] and Ponzi et al. [1], and we compared the clinical manifestations with these cases (Table 1). Fonseca et al. [19] reported various symptoms including growth and developmental delays. In this case, EEG showed abnormal focal theta semi-rhythmic activity, and there were no abnormal findings on brain MRI [19]. The patient in Ponzi et al.’s report [1] had a high birth weight of greater than the 97th percentile and hypotonia, sleep disturbance, and attention problems. This patient had normal EEG, and brain MRI revealed hypoplasia of the corpus callosum including posterior portion abnormalities of the lateral ventricles [1]. The patient in our case differed in that he was a preterm infant who had showed seizures and hypotonia since the neonatal period, which progressed to hypertonia in infancy. In addition, compared to the other cases, he had severe growth and neurodevelopmental delay, and both EEG and brain MRI showed abnormal findings. All three cases were de novo mutations with no parental influence.
Clinical manifestations of microdeletion syndrome vary widely depending on the genes included in the deleted regions of the chromosome. The present case was also difficult to diagnose due to the presence of multiple comorbidities, and a specific diagnosis was not made despite several tests, including chromosomal karyotyping in the neonatal period. However, through continuous follow-up, we were able to confirm progressive developmental and growth delay and perform chromosomal microarray analysis at an early age of 8 months. Nowadays, chromosomal microarray analysis is an essential test, and important for confirming the diagnosis of multiple congenital anomalies and neurodevelopmental disabilities during the neonatal period. Further, the present case had a large deletion that resulted in a wide array of symptoms with severe developmental and growth delay compared to previous cases. Thus, the specific diagnosis of 14q12q13.3 microdeletion syndrome in this case enabled early intervention for better management of the patient and genetic counseling for the parents.