Autosomal Recessive Malignant Infantile Osteopetrosis Associated with a TCIRG1 Mutation: A Case Report of a Neonate Presenting with Hypocalcemia in South Korea
Article information

Abstract
Osteopetrosis refers to a group of genetic skeletal disorders characterized by osteosclerosis and fragile bones. Osteopetrosis can be classified into autosomal dominant, autosomal recessive, or X-linked forms, which might differ in clinical characteristics and disease severity. Autosomal recessive osteopetrosis, also known as malignant osteopetrosis, has an earlier onset, more serious clinical symptoms, and is usually fatal. We encountered a 1-day-old girl who was born full-term via vaginal delivery, which was complicated by meconium-stained amniotic fluid, cephalo-pelvic disproportion, and nuchal cord. Routine neonatal care was provided, in addition to blood tests and chest radiography to screen for sepsis, as well as skull radiography to rule out head injuries. Initial blood tests revealed hypocalcemia, which persisted on follow-up tests the next day. Radiographic examinations revealed diffusely increased bone density and a "space alien" appearance of the skull. Based on radiographic and laboratory findings, the infantile form of osteopetrosis was suspected and genetic testing for identification of the responsible gene. Eventually, a heterozygous mutation of the T cell immune regulator 1, ATPase H+ transporting V0 subunit a3 (TCIRG1) gene (c.292C>T) was identified, making this the first reported case of neonatal-onset malignant osteopetrosis with TCIRG1 mutation in South Korea. Early-onset hypocalcemia is common and usually results from prematurity, fetal growth restriction, maternal diabetes, perinatal asphyxia, and physiologic hypoparathyroidism. However, if hypocalcemia persists, we recommend considering 'infantile of osteopetrosis' as a rare cause of neonatal hypocalcemia and performing radiographic examinations to establish the diagnosis.
INTRODUCTION
Osteopetrosis (OP) is a rare genetic disease characterized by osteosclerosis and a predisposition to fracture. OP may be classified according to clinical severity, bone marrow histology, and genetic mutation. The mode of inheritance may be autosomal recessive, autosomal dominant, and more rarely, X-linked. Autosomal dominant osteopetrosis (ADO) is the most common, and is defined as the benign adult form of OP. Patients with ADO present with mild symptoms, with onset between late childhood and adulthood. X-linked OP is very rare, and only a few cases have been reported. This form presents with classical OP symptoms, including lymphedema, immunodeficiency, localized fluid retention, and ectodermal dysplasia. Autosomal recessive malignant OP is a life-threatening disease caused by abnormalities in osteoclast differentiation and function, with an incidence rate of 1 in 250,000 births [1]. The initial clinical manifestations may be due to defective bone remodeling of the skull, and include macrocephaly, frontal bossing, and choanal stenosis, which can cause impaired nasal breathing. Hypocalcemia and secondary hyperparathyroidism may also develop due to impaired calcium homeostasis. In the case of newborns, increased bone density on radiography is a common sign of malignant OP. As the disease progresses, tetany (due to hypocalcemia), anemia or recurrent infections (due to hematopoietic failure), growth failure, and other related symptoms may develop. The most severe complication of OP is marrow insufficiency, which can be fatal before the age of 10 years [2]. Several genes have been implicated as the leading cause of autosomal recessive osteopetrosis (ARO), including T cell immune regulator 1, ATPase H+ transporting V0 subunit a3 (TCIRG1; approximately 50% of cases), chloride voltage-gated channel 7 (CLCN7; 10% to 15%), osteopetrosisassociated transmembrane protein 1 (OSTM1; 4%), and sorting nexin 10 (SNX10; 4%). When malignant OP is suspected based on radiographic findings, genetic testing should be conducted to confirm and characterize the specific functional alterations, and to determine the appropriate therapeutic strategies [3-5]. The only curative treatment for ARO is hematopoietic stem cell transplantation (HSCT), which should be performed early to achieve better outcomes.
CASE REPORT
A 1-day-old girl weighing 3,020 g was born at our hospital at 39+2 weeks gestational age by spontaneous vaginal delivery. During delivery, the amniotic fluid was moderately stained with meconium, and a nuchal cord was present. At birth, her vital signs were stable, and her Apgar scores were 8 and 9 at 1 and 5 minutes, respectively. She was subsequently admitted to the neonatal unit. On the second day of life, blood tests and chest radiography were performed to determine if the patient's meconium-stained amniotic fluid caused neonatal sepsis, while skull radiography was done to exclude severe head injuries from head molding. Complete blood count (CBC) results were nonspecific, while other biochemistry findings were as follows: serum total calcium 6.7 mg/dL (reference, 9 to 10.6), ionized calcium 0.9 mmol/L (reference, 1.07 to 1.27), phosphorus 5.0 mg/dL, blood urea nitrogen 18.2 mg/dL (reference, 6 to 20), creatinine 1.51 mg/dL (reference, 0.5 to 0.9), aspartate transaminase 125 IU/L (reference, 5 to 32), alanine aminotransferase 7 IU/L, alkaline phosphatase 126 IU/L, and creatinine kinase brain isoenzyme fraction 34.0% (reference, 0.0%). Her birth weight, length, and head circumference were 3,020 g (10th to 25th percentile), 51 cm (50th to 75th percentile), and 34.5 cm (50th to 75th percentile), respectively.
Her mother was a healthy 22-year-old unmarried woman with unremarkable family and social history, except for a history of alcohol (5 oz per day) during the 1st trimester. Details of her father were not provided.
Radiographic findings revealed spina bifida at T2, bilateral aplasia of the 12th ribs, absence of corticomedullary junctions in the humerus, and a “space alien” appearance with osteosclerosis of the skull base. Additional radiography of the lower extremities was performed for evaluation of bone density (Figure 1). She was fed with cow’s milk formula without any problems. No seizures, or respiratory difficulties were noted. She was admitted to the neonatal intensive care unit (NICU) for further evaluation, and calcium supplementation was administered on the 2nd day of life.
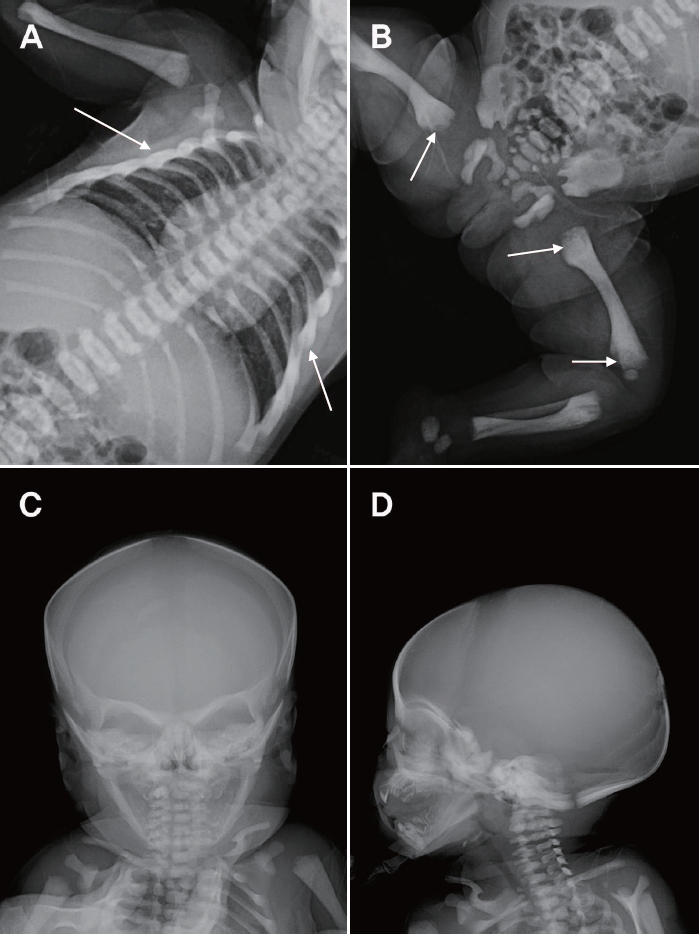
Radiographic findings. (A, B, C, D) Diffusely increased bone density is seen on all images. (A, B) Arrows show sclerotic bone changes. The “space alien” appearance of the skull is shown in (C).
Additional biochemistry investigations performed on the 3rd day of life showed a parathyroid hormone (PTH) level of 308 pg/mL (reference, 9 to 54), and 25-OH vitamin D level of 5.71 ng/mL (reference, 30 to 50). Her hypocalcemia, vitamin D deficiency, and secondary hypoparathyroidism were treated with intravenous calcium gluconate (225 mg once a day, Choongwae Pharm, Seoul, Korea) and vitamin D (1,200 IU). On the 5th day of life, her serum total calcium level normalized (8.7 mg/dL), and the dose of elemental calcium was reduced (75 mg/kg/day, calcium carbonate, Cicibon Tab, Huons, Seoul, Korea). Besides the persistently elevated PTH level (473 pg/mL), those of serum total calcium (9.3 mg/dL), and 25-OH vitamin D (23.96 ng/mL) normalized after 2 weeks of calcium supplementation on the 15th day of life. Vitamin D supplementation was continued at a reduced dose (400 IU) and calcitriol (0.25 μg/day, Bonky Soft Cap, Yuyu Pharmaceutical, Seoul, Korea) was added as a temporary adjuvant therapy to improve gastrointestinal absorption of supplemental calcium. These were maintained until HSCT was performed.
At 3 weeks of life, follow-up CBC results were as follows: hemoglobin 9.0 g/dL, hematocrit 27.7%, white blood cell count 8,900/μL, and platelet count 201×103/μL, with differential counts of 4% metamyelocytes, 4% band neutrophils, 25% segmental neutrophils, 62% lymphocytes, and 4% monocytes, suggesting the progression of bone marrow suppression. Based on these finding, a red blood cell transfusion (10 mg/kg/dose) was initiated. A blood sample was sent to Samsung Medical Center (SMC) for molecular genetic analysis of OP, and a heterozygous mutation in the TCIRG1 gene (c292C>T) was found. While hospitalized at our NICU, she did not show any respiratory difficulties, and showed no signs of visual and hearing impairment. She was discharged on the 34th day of life, and was referred to SMC for HSCT.
DISCUSSION
OP is a rare genetic disorder characterized by partial or complete loss of osteoclast function, leading to bone resorption disorders. Most forms of OP are osteoclast rich, and are caused by impaired gene products involved in acidification. Acid secretion is dependent on two key molecules that promote proton transport: the proton pump vacuolar ATPase (V-ATPase) and the chloridespecific ion channel, chloride channel 7 (CIC-7) [1,6,9]. Mutations in V-ATPase a3 subunit (TCIRG1) and CIC-7 (CLCN-7) genes produce severe malignant OP phenotypes in both humans and mice [1,6-8]. TCIRG1 mutations are responsible for more than 50% of ARO-affected individuals [1,7-10]. On the other hand, mutations in CLCN-7, which plays a crucial role in lysosomal acidification, lead to severe neuronal storage and neurodegeneration in the central nervous system (CNS) and retina in a subset of ARO patients [1,11,12]. Some types of single allelic mutation in CNCN-7 can also cause the benign form of ADO in adults. Recently, mutations in the SNX10 gene have been identified [2,13]. Osteoclast-poor OP has been reported, but is rare. In this form of OP, osteoclasts may be absent, and genetic defects have been reported to reside in either the receptor activator of nuclear factor-κB ligand (RANKL) or RANK gene, which encodes for critical factors of preosteoclast fusion. Other rare forms of OP, may be caused by mutations in CAII, OSTM1, nuclear factor κB essential modulator (NEMO), pleckstrin homology and RUN domain containing M1 (PLEKHM1), KINDLIN3, and solute carrier family 29 member 3 (SLC29A3), which impair osteoclast activity through different mechanisms (Figure 2). Such genetic variability results in a wide phenotypic heteroeneity among patients, ranging from asymptomatic to fatal diseases in infancy. X-linked OP is also known as osteopetrosis-lymphedema-ectodermal dysplasia anhidrotic with immunodeficiency (OL-EDA-ID), an acronym derived from each of the major features of the disorder. It is caused by mutations in nuclear factor κB (NF-κB), which is an essential modulator in hemizygous male patients [14].
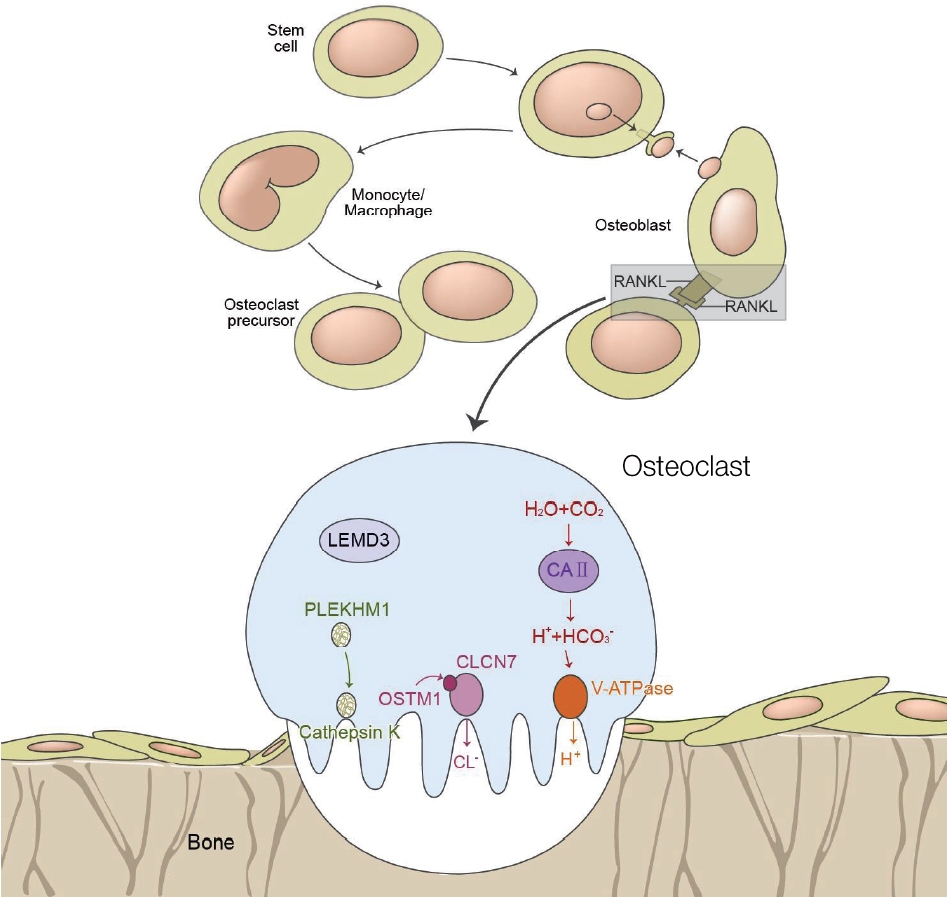
Differentiation of osteoclasts and representation of the current pathogenetic model of osteopetrosis. Adapted from Stark et al.[1] and Tolar et al.[15], with permission from Massachusetts Medical Society. Abbreviations: RANKL, receptor activator of nuclear factor-κB ligand; LEMD3, LEM domain-containing protein 3; PLEKHM1, pleckstrin homology and RUN domain containing M1; CLCN7, chloride voltage-gated channel 7; OSTM1, osteopetrosis-associated transmembrane protein 1; V-ATPase, vacuolar ATPase.
Despite the increased bone mass and osteosclerosis on radiography, osteopetrotic bones are predisposed to fractures. Disruption of bone remodeling can lead to skeletal and dental deformities, and can impair mineral homeostasis. In addition, expansion of bones into the marrow cavity and cranial nerve foramina can compromise hematopoiesis and neurological functions, respectively.
The clinical presentations of patients with malignant OP are heterogeneous; therefore, it is difficult to determine a specific treatment strategy for each situation. Once a radiological and clinical diagnosis of malignant OP is made, the type of mutation, age at presentation, and presence of risk factors should be considered. Genetic testing is crucial to characterize the phenotype and inheritance pattern of OP, and to determine the need for HSCT.
Although HSCT is an option for specific mutations intrinsic to osteoclasts, there are several contraindications, including extrinsic osteoclast defects (RANKL gene mutations), neurodegenerative forms of OP (OSTM1 gene mutation), and mutations involving CLCN7 gene. Pharmacological treatments, such as corticosteroids, vitamin D, calcium supplementation, PTH, and gamma-interferon, are only temporary measures and cannot substitute for HSCT.
In the present case, the patient was hospitalized to receive supportive treatment for hypocalcemia, which was detected by chance during screening tests. She also demonstrated reduced 25-OH vitamin D, and increased PTH levels due to secondary hypoparathyroidism as well as increased bone density of the spine, long bones, and skull on radiography. As such, calcium, calcitriol, and vitamin D supplements were prescribed. A generalized increase in bone density of the spine and the upper and lower extremities, as well as the "space alien" appearance of the skull, were the diagnostic clues for malignant OP (Figure 1C). While serum calcium levels improved after 1 week of calcium supplementation, calcium and calcitriol administration was maintained until HSCT was performed.
The patient was diagnosed with a TCIRG1 mutation (NM_006019.4, c.292C>T, p.Gln98Ter). Such mutation typically involves a single nucleotide variant, in which glutamine (Gln) is substituted with a stop codon (Ter), and is associated with pathogenic OP. Patients with TCIRG1 mutation (OMIM #2597 00; OPTB1; vacuolar H+-ATPase) present with classical malignant infantile osteopetrosis (MIOP) [4]. Milder forms have rarely been reported [3,5,15,16]. Clinical manifestations include growth retardation, hypocalcemia, hematologic impairment, visual impairment, and CNS symptoms. MIOP is one of the TCIRG1-related forms of that is indicated for HSCT. Early transplantation may associate with better outcomes.
In our patient, allogenic peripheral blood stem cell transplantation (allo-PBSCT) was performed at 5 months of age with regular follow-ups thereafter. Her serum calcium levels remained within normal ranges following allo-PBSCT. In addition to facilitating the definitive diagnosis and management of OP, genetic analyses can be useful for couples with recurrent miscarriage due to single gene mutations or chromosomal aberrations. Preimplantation genetic diagnosis (PGD), which is performed on chorionic villus or amniocentesis samples by direct sequencing [17], can help in achieving a normal pregnancy by early detection of affected embryos.
The optimal care of malignant OP patients requires the involvement of a multidisciplinary team. In our case, an ophthalmologist and otolaryngologist were consulted, despite the absence of visual or hearing defects at the time of OP diagnosis. The multidisciplinary team may include endocrinologists, ophthalmologists, geneticists, orthopedic surgeons, neurologists, neurosurgeons, dentists, otolaryngologists, hematologists, infectious disease specialists, nephrologists, pain management specialists, and developmental pediatricians [18].
Ultrasonographic evaluations were performed to exclude hepatosplenomegaly due to extramedullary hematopoiesis following bone marrow failure. However, no organomegaly was observed, owing to the very young age at diagnosis. Sclerotic bones are not a common initial finding in young MIOP patients; instead, more generalized clinical manifestations, such as jaundice, infection, and respiratory difficulty due to choanal atresia, predominate as the presenting symptoms. Specific radiographic findings usually appear later, which can lead to delayed diagnosis. To date, two cases of ARO have been reported in Korea. One of them was a neonate who presented with thrombocytopenia due to a mutation of CLCN7 mutation. The patient eventually died of to sepsis, but his mother, who had experienced repeated abortions and stillbirths, underwent PGD for her next pregnancy and gave birth to a healthy baby. The other patient presented with cytomegalovirus infection with a CLCN7 mutation being the causative etiology.
In summary, we encountered a 2-day-old girl who presented with hypocalcemia without seizures and generalized increased bone density on chest and skull radiographies, which led to a diagnosis of MIOP. Genetic analysis revealed a TCIRG1 (c.292C>T). The patient was referred for HSCT, and has been regularly followed up thereafter. This is the first reported case of a TCIRG1 mutation causing neonatal-onset malignant OP in South Korea.
Notes
Ethical statement
This study was approved by the Institutional Review Board of Sung-Ae Medical Foundation (IRB No. SA2021-03). Informed consent was waived by the board due to a retrospective nature of our study.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Author contributions
Conception or design: Y.K.O., Y.J.S.
Acquisition, analysis, or interpretation of data: Y.K.O., K.E.C., Y.J.S.. E.R.K., J.Y.K., M.S.K., S.Y.C., D.K.J.
Drafting the work or revising: Y.K.O, K.E.C, Y.J.S. E.R.K.
Final approval of the manuscript: Y.J.S.
Acknowledgements
None