A Novel COL4A1 Mutation in a Neonate with Intrauterine Intraventricular Hemorrhage and Porencephaly
Article information

Abstract
Collagen type IV alpha 1 (COL4A1) plays an important role in construction of the basement membranes of all human tissues, especially vessels. Mutations in COL4A1 lead to various multisystemic dysfunctions, including hereditary porencephaly, hemorrhagic stroke, hemiplegia, cerebral small vessel disease, and nephropathy. In this study, we describe a neonatal case featuring a novel de novo COL4A1 mutation, manifesting as fetal intraventricular hemorrhage and porencephaly. This patient is one of the youngest to have been diagnosed with the most severe phenotype. Our experience may assist clinicians in the diagnosis and management of this extremely rare genetic condition.
INTRODUCTION
Fetal porencephaly is classified into two subtypes: developmental and encephaloclastic. The former represents the primary failure of neuronal development and migration, and the latter develops when an environmental or genetic insult destroys a normally developed brain. Congenital encephaloclastic porencephaly has many different causes; of these, a defect in collagen type IV alpha 1 (COL4A1) leads to fragile blood vessels with fetal hemorrhagic strokes [1]. COL4A1 is located on chromosome 13q34, and encodes the COL4A1 [2]. COL4A1, which is expressed in all tissues, is a crucial component of basement membrane proteins. In particular, it is an integral protein in angiogenesis, regulating blood vessel tone, cohesiveness of the basal membrane, and endothelial function [3]. Defects in COL4A1 lead to multisystemic abnormalities affecting the brain, eyes, kidneys, muscle, and other organs [4,5]. In particular, the affected fetus may present with intrauterine intraventricular hemorrhage and subsequent porencephaly or schizencephaly.
In this report, we describe a male newborn affected by severe fetal porencephaly and a heterozygous mutation. To the best of our knowledge, this is the youngest child with a reported COL4A1 defect.
CASE REPORT
A male newborn was born vaginally at 38+3 weeks’ gestation to a 31-year-old mother. At 21 weeks’ gestation, he was suspected to have intraventricular hemorrhage, porencephaly, and bilateral cataracts, identified during prenatal ultrasonography (Figure 1). There was no family history of congenital anomaly and systemic illness including his parents and two healthy brothers.
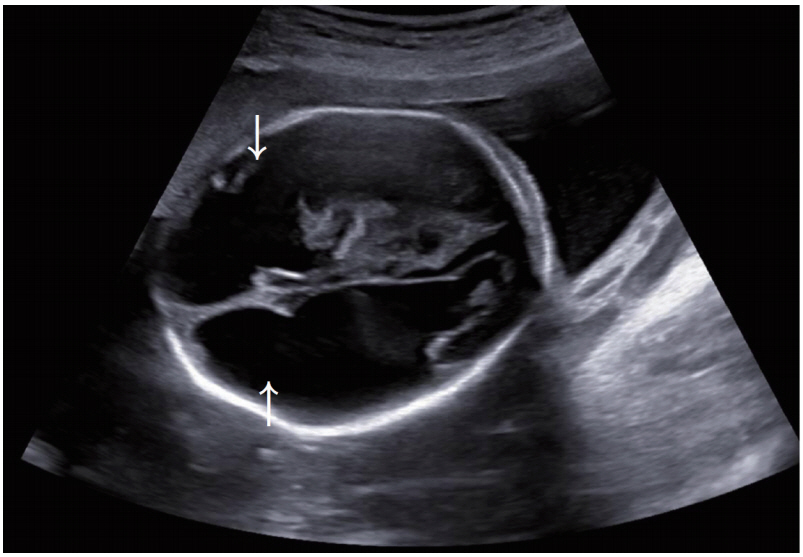
Bilateral frontal porencephaly on fetal sonography at 21 weeks of gestation (white arrows).
His birth weight, birth length, and head circumference were 2,180 g (<3rd percentile), 45 cm (<3rd percentile), and 31 cm (<3rd percentile), respectively. He had intrauterine growth retardation, but there were no complications during pregnancy. At birth, his Apgar scores were 7 and 9 at 1 and 5 minutes, respectively. On physical examination, microcephaly, micrognathia, facial purpura, and bilateral clubfoot were noted. On ophthalmological investigation, bilateral cataracts were found (Figure 2). His hemoglobin level was 12.2 g/dL and normocytic normochromic anemia without fragmented red blood cells (RBCs) were identified on a peripheral blood smear. His platelet count, prothrombin time, and activated partial thromboplastin time were normal. Tests for toxoplasmosis, rubella, cytomegalovirus infection, and herpes infection were all negative. Brain sonography at day 1 revealed proportionally dilated bilateral lateral ventricles that were more dilated anteriorly (Figure 3A). Brain magnetic resonance imaging at day 1 revealed bilateral ventriculomegaly communicating with extensive porencephaly in the bilateral frontal region (Figure 3B). There was marked cortical thinning with almost no discernible cortical mantle in the frontal region. In addition, evidence of previous hemorrhage with hemosiderin deposition along the margins of the ventricles and bilateral choroid plexi was noted (Figure 3C). Magnetic resonance angiography revealed slightly attenuated intracranial vessels without significant tortuosity or abnormal dilatation. The results of electrocardiogram, echocardiogram, and kidney ultrasonography were normal. His karyotype was 46, XY. The hospital course was uneventful, and he was discharged at 10 days of life (DOL).
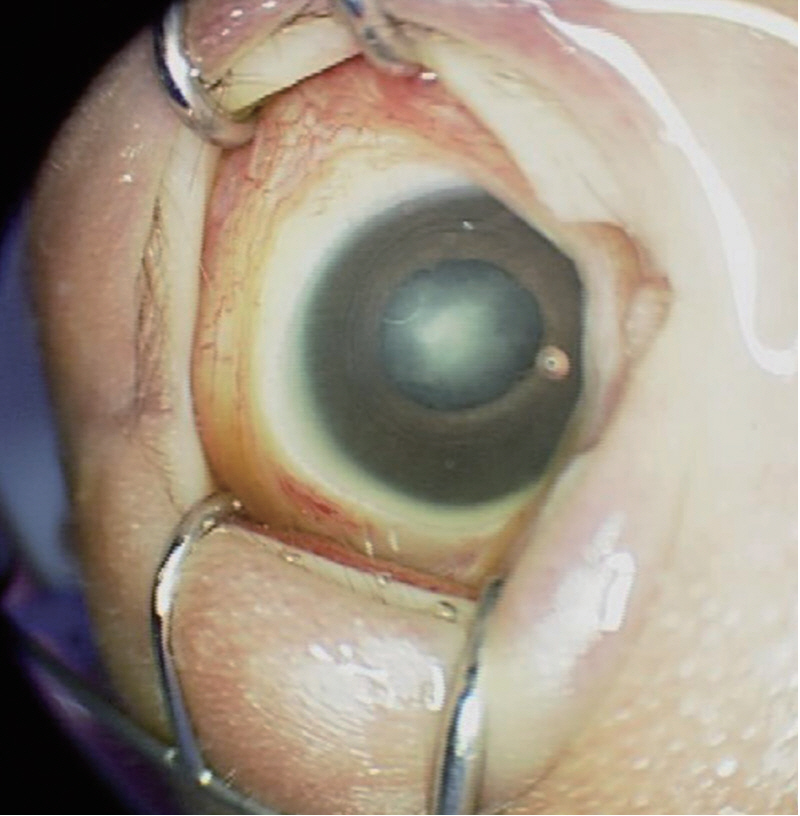
Right cataract of the patient.
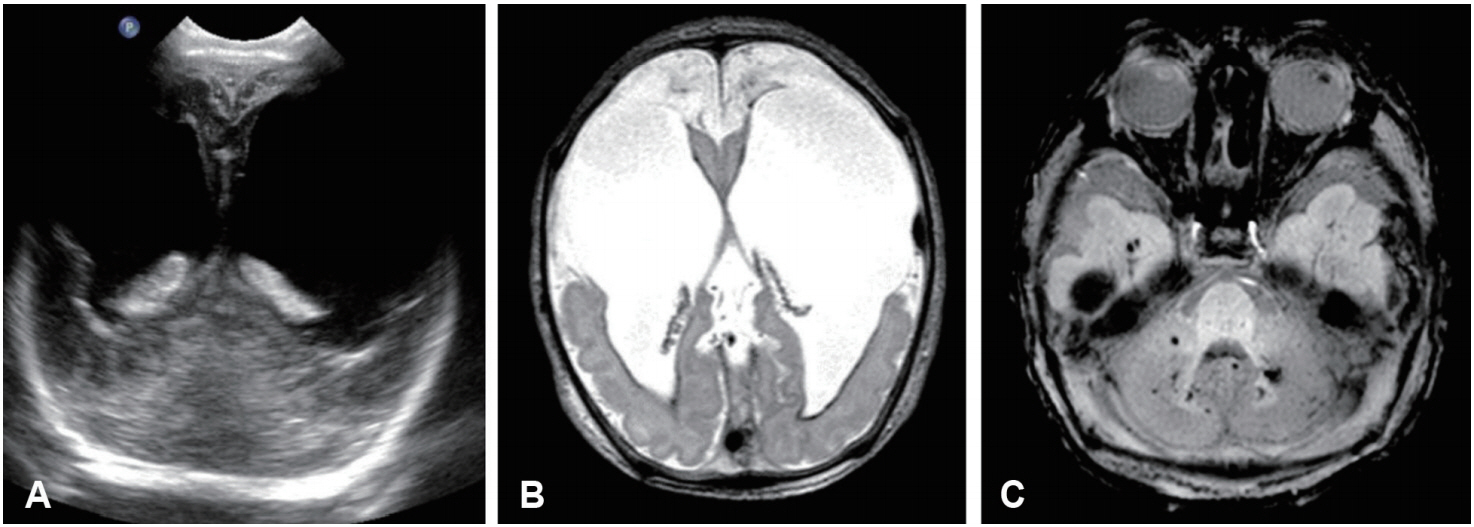
Images of a microcephalic newborn boy with collagen type IV alpha 1 (COL4A1) mutation. (A) Cranial ultrasonography revealed dysmorphic enlarged lateral ventricles. (B) Axial T2-weighted imaging of the brain revealed bilateral dilated lateral ventricles communicating with extensive frontal porencephaly. Marked thinning of the cortical mantle was noted in the bilateral frontal region. T2 dark signal intensity suggesting hemosiderin deposition was observed along the linings of the lateral ventricles. (C) Axial susceptibility weighted imaging revealed multiple foci of susceptibility in the cerebellum suggesting microhemorrhages.
At 2 months of age, he was admitted to our hospital because of an enterovirus infection. Hemolytic anemia, as indicated by a hemoglobin level of 6.3 g/dL with fragmented RBCs, was noted. In addition, his reticulocyte, haptoglobin, and plasma hemoglobin levels were 15.78%, <7.8 mg/dL, and 8.9 mg/dL, respectively. Direct and indirect Coombs test were negative. There was no definite evidence of additional hemorrhage on the brain ultrasonography at admission. He received a 10 mL/kg RBC transfusion, and his hemoglobin level at discharge was 7.6 g/dL. No additional transfusion was performed.
Partial exome sequencing of 4,813 Online Mendelian Inheritance in Man (OMIM) genes was performed using peripheral leukocytes at 10 DOL under the suspicion of COL4A1 mutation. Exomes were captured using the TruSight One Panel (Illumina Inc., San Diego, CA, USA), which enriches a 12 Mb region spanning 4,813 genes. Sequencing was performed using the Next Seq platform (Illumina Inc.). Sequence reads were aligned to the reference genome, hg19, using BWA (v.0.7.12, MEM algorithm). The mean depth of coverage was 90× (>10×=98%). The heteroFigure zygous novel COL4A1 mutation c.2645G>T (p.Gly882Val) was identified (Figure 4). In silico prediction tools, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) and SIFT (http://sift.jcvi.org), were employed. This variant was classified as pathogenic according to the American College of Medical Genetics and Genomics [6]. Neither of his parents carried the mutation.

Sequence chromatograms revealed that a single heterozygous nucleotide change, c.2645G>T (p.Gly882Val), was present (A) in the patient, but not in his (B) father or (C) mother.
At 9 months of age, phacoemulsification cataract extraction with posterior chamber lens implantation was performed for bilateral congenital cataracts. At 12 months of age, his weight, height, and head circumference were 10th percentile, 10th percentile, and under the 3rd percentile, respectively. He exhibited profound developmental delay. He could only roll supine to a prone position and make vowel-like noises. He was able to eat formula milk orally without a feeding tube, although intermittent aspiration occurred. On the Bayley Scale of Infant Development-II, his mental and psychomotor development indices were 8 and 15, respectively, indicating severe developmental cognitive and motor delay. He was diagnosed with epilepsy and took anticonvulsants. Hemolytic anemia was no longer observed after enterovirus infection. Microscopic hematuria was observed but no specific findings were found during renal sonography.
DISCUSSION
As a component of type IV collagen, COL4A1 encodes its alpha 1 chain. This chain combines with another alpha 1 chain and the alpha 2 chain to form complete type IV collagen alpha 1-1-2 heterotrimers. These molecules attach to each other to form a complex protein network, which is important for basement membrane interactions and cell migration, proliferation, maturation, and survival.
The spectrum of COL4A1 defect-related disorders includes cerebral small-vessel disease, variably associated eye defects (Axenfeld-Rieger anomaly, cataract, retinal arterial tortuosity, retinal anterior segment dysgenesis), and systemic disorders (kidney involvement, muscle cramps, Raynaud phenomenon, cardiac arrhythmia, and hemolytic anemia) [7,8]. HANAC (hereditary angiopathy with nephropathy, aneurysms, and muscle cramps) syndrome is usually associated with asymptomatic cerebral small vessel disease [9,10].
COL4A1 mutations represent an autosomal dominant cause of hereditary porencephaly, most often caused by germinal matrix hemorrhage leading to deep venous infarction with subsequent tissue necrosis and porencephalic cavitation [1,10]. The incidence of COL4A1 mutations among 61 pediatric patients with porencephaly and schizencephaly was 21% in an analysis conducted by Yoneda et al. [5]. In another cohort, 183 patients with porencephaly or infantile hemorrhage were selected, and the researchers identified 21 COL4A1 and 3 COL4A2 pathogenic mutations (13%) [4].
In our case, the patient exhibited porencephaly, congenital cataract, and hemolytic anemia, which are the symptoms of COL4A1-related disorders. Porencephaly, a typical manifestation of COL4A1 mutation, is characterized by cystic cavities that communicate with ventricles and that are thought to arise from germinal matrixhemorrhage. Previous studies reported a potential link between congenital cataracts and COL4A1 mutation [11,12]. COL4A1 expression was measured in human embryonic and adult lens capsules. The differentiation of mesenchymal cells in the cornea and the formation of an anterior chamber depend on signals controlled by transcription factors that are specifically expressed in mesenchymal cells and on inductive signals from the lens. Mutations in COL4A1 may disrupt some lenticular signaling in the direction of mesenchymal cells [13,14]. The hemolytic anemia reported in a previous study was also observed in the present case; however, the exact cause has not yet been identified, and other studies suggested that this symptom is less relevant to COL4A1 mutation [5,15]. The patient also exhibited developmental delay, resulting from characteristic porencephaly. The neurologic patterns associated with COL4A1 mutation comprise a typical severe presentation and a spectrum of less common phenotypes. The typical severe phenotype with porencephaly was defined by severe developmental delay, intellectual and behavioral difficulties, microcephaly, and motor abnormalities on neurologic examination. Even in the absence of any cerebrovascular hemorrhage, some patients with COL4A1 mutations display leukoencephalopathy, calcification, or cerebral microbleeding. These silent defects might cause physical disability and global developmental delay [16].
COL4A1 mutations are passed on as autosomal dominant mutations, and most cases have been sporadic. Therefore, the risk for parents to have another affected child is less than 1%. However, because of the possibility of germline mosaicism, prenatal genetic testing for future pregnancy might be required [17].
To the best of our knowledge, our case, which was diagnosed at 57 DOL, is the earliest diagnosed COL4A1 mutation case on record. After diagnosis, we were able to provide more accurate information and more systematic clinical follow-up and treatment. In this disease, avoiding additional head trauma and anticoagulant exposure may help to reduce the risk of additional intracranial hemorrhage with personalized treatment for individual symptoms [17]. Therefore, after a rapid diagnosis, parental education was offered to reduce additional head trauma. It also provided an opportunity for prenatal genetic counseling related to the future pregnancy. In newborns with multiple congenital anomalies such as porencephaly, cataracts, and hemolytic anemia, it is important to consider COL4A1 mutation and conduct early genetic testing. In addition, we suggest careful antenatal genetic testing for COL4A1 mutations in fetuses with intrauterine brain hemorrhage, porencephaly, or schizencephaly given the currently undefined related phenotype.
Notes
Ethical statement
The Institutional Review Board (IRB) approval for the study was received in June 2019. Informed consent was waived by the board. Retrospective data collection was approved by the IRB of Asan Medical Center (IRB No. 2019-0745).
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Author contributions
Conception or design: E.J., B.H.L.
Acquisition, analysis, or interpretation of data: J.N., E.J., A.Y.J., B.H.L.
Drafting the work or revising: J.N., E.J., A.Y.J., B.H.L., B.S.L., E.A.R.K., K.S.K.
Final approval of the manuscript: J.N., E.J., A.Y.J., B.H.L., B.S.L., E.A.R.K., K.S.K.
Acknowledgements
None